SAMtools
Download and Install
Download tool packages samtools from [original website] of the study [1]. The following processes take version 0.1.19 as a example to implement the operating samtools.
In the command line mode to use samtools, move the compressed file (named: samtools-0.1.19.tar.bz2) to the path where you want to place and execute. The example takes /home/(user)/桌面 as a example.
$ mv ./samtools-0.1.19.tar.bz2 /home/JKW/桌面/Extract (unzip) the compressed file by the following instructions;
$ tar -xvf ./samtools-0.1.19.tar.bz2Move into the folder by cd instruction as following;
$ cd ./samtools-0.1.19try to compile all necessary files for executing samtools by instruction make;
$ makeIf there is error while compiling samtools files, for example, lost of curses.h , it represents that you need other resources (libraries) for completing installing samtools. The following instruction is needed to solve above problems.
$ sudo yum install zlib-devel ncurses-devel ncursesAfter installing the above necessary files prepared for samtools, you would re-enter the following instruction;
$ makeOnce complete compiled processes, the executable file named samtools is generated. Also, the folder bcftools containing executable file is generated. And both these could further be used for downstream analysis.
Operations
The following processes take the previous file (named: lambda.sam) generated from bowtie2 as a example.
The following processes are to find SNPs/INDELs for the example sequence with helps of samtools and bcftools.
In general. the dataset generated from next-generation sequences is quite large. For the purpose of long-termed storage, the first thing is to convert the SAM file into BAM file. The BAM file is a binary format corresponding to the SAM file text. We could use samtools to achieve that. (assume: lambda.sam is located on the desktop and samtools package is also located on the desktop)
$ cd /home/JKW/桌面/ $ ./samtools-0.1.19/samtools view -bS ./lambda.sam > ./lambda.bamFor a quick searching of reads from reference genome, a sorted dataset is necessary for this purpose. We could use samtools with parameter sort to achieve that.
$ ./samtools-0.1.19/samtools sort ./lambda.bam ./lambda.sortedNext, we could use samtools with parameters mpileup and bcftools to generate variant calls in BCF format.
$ ./samtools-0.1.19/samtools mpileup -uf ./lambda_virus.fa ./lambda.sorted.bam | ./samtools-0.1.19/bcftools/bcftools view -bvcg - > ./lambda.raw.bcfThen, we use use bcftools with parameter view to take a look into variants as the following instruction.
$ ./samtools-0.1.19/bcftools/bcftools view ./lambda.raw.bcf > ./result.txtThe following is the result executing above instructions;
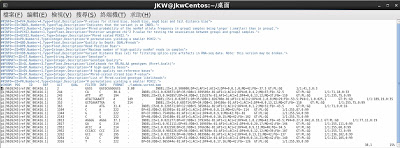
Reference
- Heng Li, Bob Handsaker, Alec Wysoker, Tim Fennell, Jue Ruan et al. (2009) The Sequence Alignment/Map format and SAMtools. BIOINFORMATICS Vol. 25 no. 16, pages 2078–2079
- The SAM/BAM Format Speci.cation Working Group. (2013) Sequence Alignment/Map Format Specication.
- 有勁的生物與資訊. 教學-於 Window 7 平台下使用 samtools 尋找 SNPs / InsertAndDeletions. (2013)