SAM files
introduction
SAMtools are a package implementing various functions for post-processing alignments in SAM format, for example, indexing, sorting, variant caller and alignment viewer.
Samtools is designed to support all sequence types and aligner and to create a well-defined interface between alignment and downstream alayses, such as assembly.
The following is to introduce SAM format:
- SAM file is composed of several groups in the format of one header section ("@" is at front) and one alignment section . All lines are TAB ('\t') delimited.
- SAM file is consisted of (1)eleven mandatory fields and (2)a variable number of optional fields.
Data infomation
- Eleven mandatory fields are the following [2]
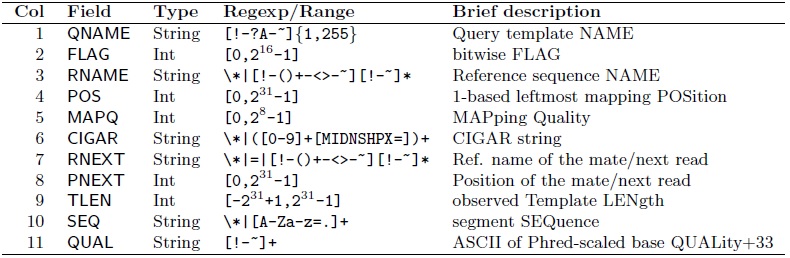
- Each item could be further discussed more detailed, for example, the FLAG field [2] as the following :

- One variable number of optional field is consisted of the following items:
TAG:TYPE:VALUE- TAG: two characters string matching /[A-Za-z][A-Za-z0-9]/
- TYPE: single case-sensitive letter defining the VALUE format.
- VALUE: the description

Reference
- Heng Li, Bob Handsaker, Alec Wysoker, Tim Fennell, Jue Ruan et al. (2009) The Sequence Alignment/Map format and SAMtools. BIOINFORMATICS Vol. 25 no. 16, pages 2078–2079
- The SAM/BAM Format Speci.cation Working Group. (2013) Sequence Alignment/Map Format Specication.
- 有勁的生物與資訊. 教學-於 Window 7 平台下使用 samtools 尋找 SNPs / InsertAndDeletions. (2013)