Linux 下建立本機/遠端資料庫及 BLAST 操作
- 環境:Linux-2.6.32-358.14.1.el6.i686, CentOS-6.4
架設與操作 local blast suites (以 CentOS 6.4-i686 為例)

安裝步驟
- 安裝網址:ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST
- 直接安裝檔:以 Fedora、CentOS 等系列,檔名以 .rpm 為主。
- 包裝套件檔:檔名以 .tar.gz 為主。
- 差異:下載時請注意 linux kernel 版本,位元版本。本說明檔以 Linux 2.6.32-358.14.1.el6.i686 及 CentOS 6.4 為測試對象,安裝檔案以直接安裝(檔名:ncbi-blast-2.2.28+-1.i686.rpm)為測試對象。包裝套件與直接安裝的差異僅為環境指令的搜尋路徑差異,即直接安裝已將必要的指令都鍵入尋找路徑中,可以直接由 command 下執行,而不用指定執行檔案的路徑。
取得公共序列資料庫與待測試的資料庫
- 公共序列資料庫:可以參考 NCBI 的人類公共序列資料庫(ftp://ftp.ncbi.nlm.nih.gov/refseq/H_sapiens/mRNA_Prot/), 內含有 DNA sequences、RNA sequences(in DNA format) 與 protein sequences。
- 待測試的 genome:以 yeast chromosome 1 為例(可以參考載點 http://downloads.yeastgenome.org/sequence/S288C_reference/chromosomes/fasta/),待測試資料僅需要盡量完整的序列資訊即可,可以為 DNAs 或是 proteins。
BLAST 建構 terminal databases
- 建構 DNAs/RNAs databases
$ makeblastdb –in FASTA –dbtype nucl
舉建立 refseqgene1.gnome.fna 為例,而其他建立核酸序列的方式皆相同
$ makeblastdb –in ./refseqgene1.gnome.fna –dbtype nucl
如下圖建構完成的結果
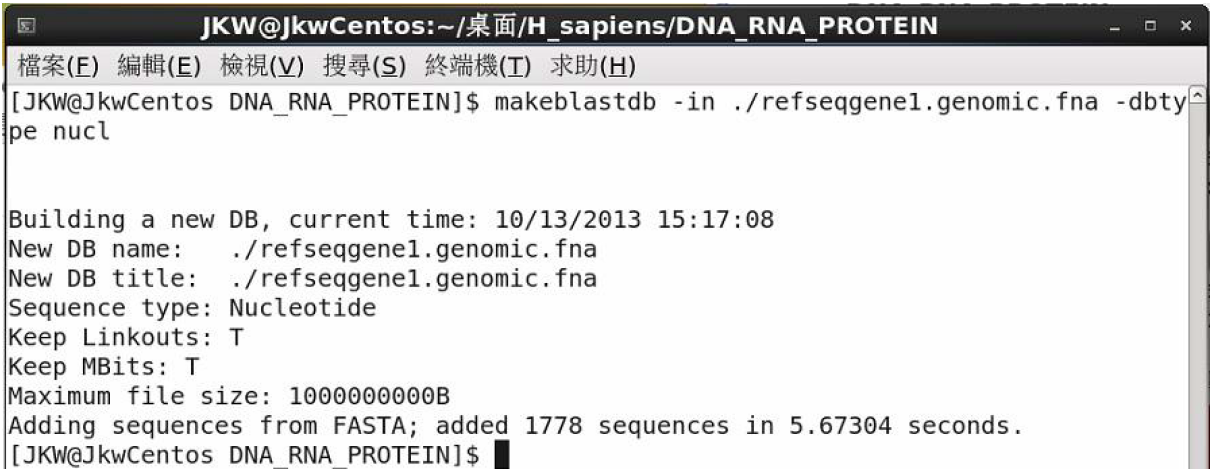
完成後,會在建立序列的同一資料夾下產生另外三個資料庫相關檔案,如下
refseqgene1.genomic.fna
refseqgene1.genomic.fna.nhr
refseqgene1.genomic.fna.nin
refseqgene1.genomic.fna.nsq
- 建立 proteins databases
$ makeblastdb –in FASTA –dbtype prot
舉建立 human.protein.faa 為例,而其他建立核酸序列的方式皆相同
$ makeblastdb –in ./human.protein.faa –dbtype prot
如下圖,建立資料庫完成後的結果;
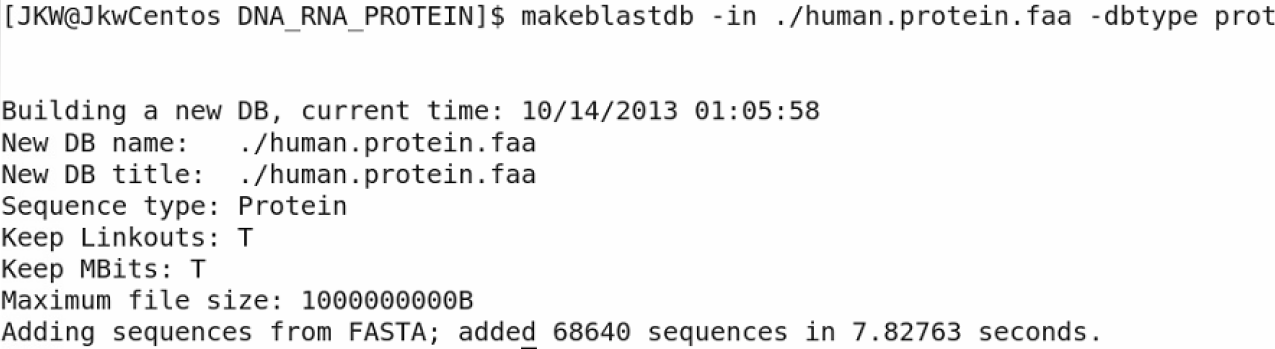
完成後,會在建立序列的同一資料夾下產生另外三個資料庫相關檔案,如下
human.protein.faa
human.protein.faa.phr
human.protein.faa.pin
human.protein.faa.psq
Blast 操作
- BLAST 操作 1 與結果:blastn
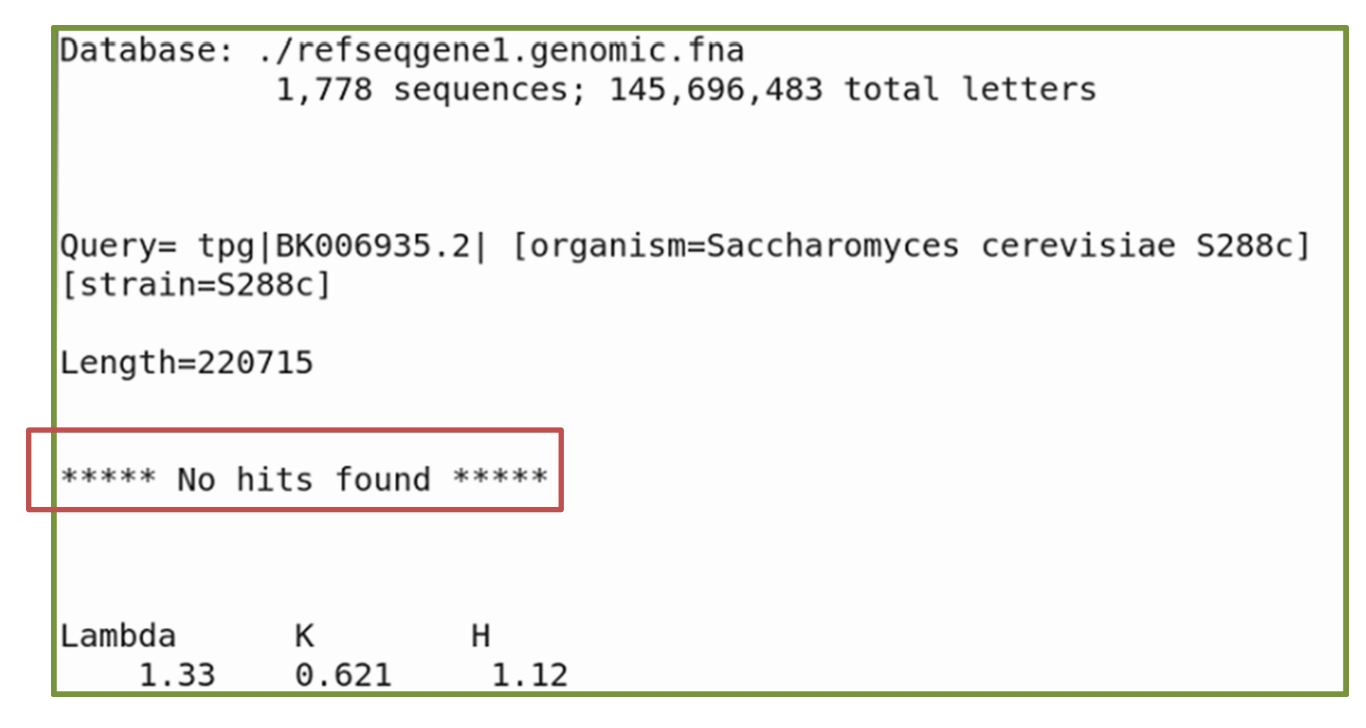
若將 yeast chromosome I 與 human DNA sequences 進行 alignment (refseqgene1.gnome.fna、refseqgene2.gnome.fna 與 refseqgene3.gnome.fna),都因為比對分數太低而沒有找到合適的序列結果 (no hits),以下舉查詢 refseqgene1.gnome.fna 為例,執行指令:
$ blastn –db ./refseqgene1.gnome.fna –query ./chr01.fsa –out blastn_out –evalue 0.000001 –num_threads 2
- blastn:從架設與操作 local blast suites 圖可知,blastn 為查詢 command,查詢與搜尋皆為 nucleotides 類型。
- –db 為選擇哪一個資料庫。
- –query 為輸入來查詢的全部序列。
- –out 後接輸出的檔案的名稱與路徑。
- –evalue 為給了一段序列後,搜尋資料庫下期望能找到 (hit) 序列的數目,代表著期望值。此值代表序列的相似度,當要搜尋一條序列時,希望能越精確越佳,所以越低越好。而可以將 evalue 當作是篩選標準。
- –num_threads 為使用多少顆處理器來進行比對,此和比對的速度有關,通常不會設定超過自己硬體最大的處理器數目。
執行結果如下:
因為 yeast chromosome I 的序列來比對目前已知 human DNA 的序列,比對後發現沒有相似(分數太低)的序列,因此為 “ No hits found. “。而不僅是 refseqgene1.gnome.fna、refseqgene2.gnome.fna 與 refseqgene3.gnome.fna 等亦是。
- BLAST 操作 2 與結果:blastx
將 yeast chromosome I 的序列來和 blast database 中的 protein databases 進行比對,當輸入序列為核酸時,會將此序列轉換成 protein sequences,共有六種,然後再利用此轉譯完成的蛋白質序列分別和 protein databases 進行比對,以下利用 yeast chromosome I sequence 和 human protein databases 進行比對,執行指令:
$blastx –db ./human.protein.faa –query ./chr01.fsa –out blastx_prot_out –evalue 0.000001 –num_threads 2
指令如下圖:

而相關的參數說明可以參考上「BLAST 操作 1 與結果:blastn」的說明;執行結果說明如下:
當用 yeast chromosome I 與 human.protein.faa 進行 blastx 比對時,是利用轉譯後的蛋白質來進行蛋白質資料庫比對,因此輸入的序列若是真核,需要考慮 splicing 的問題。
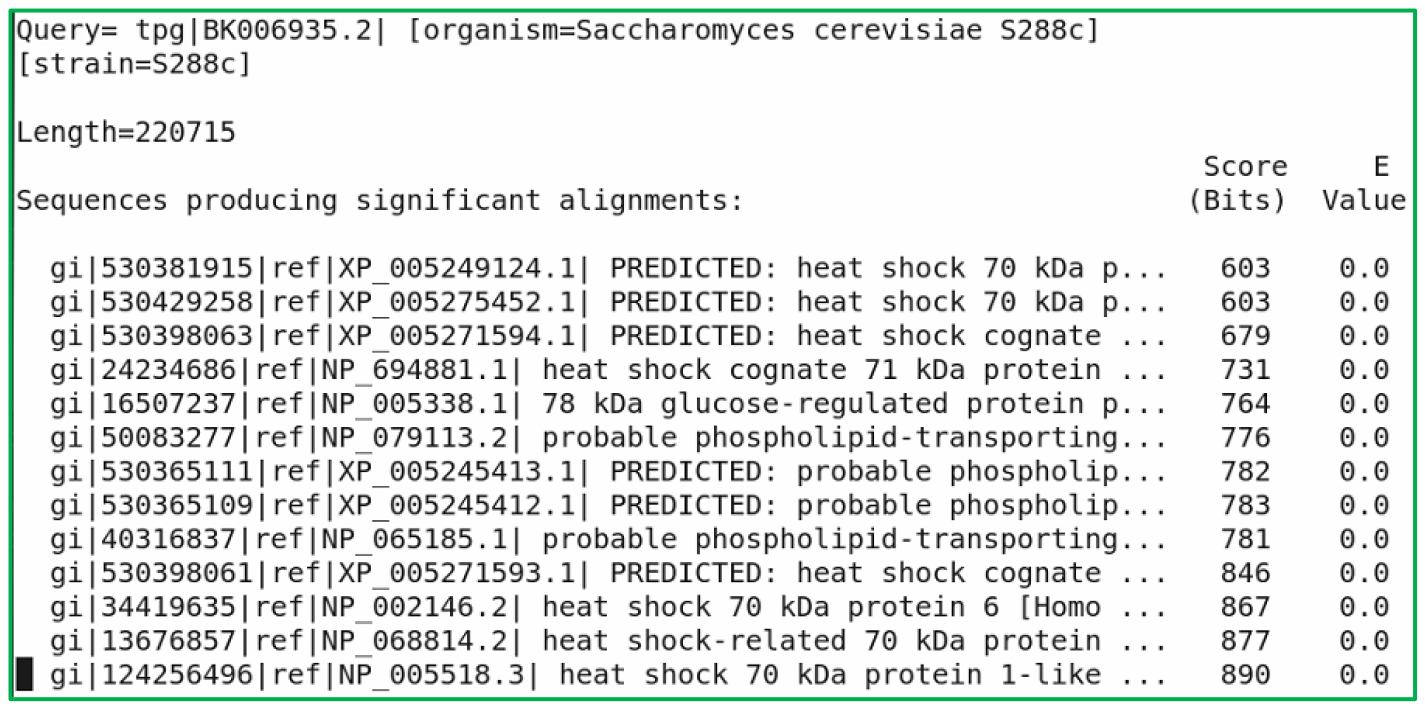
- 可以看出在 yeast chromosome I 中和 human 有許多高度相似的蛋白質序列,因此當核酸序列轉成蛋白質來比較資料庫後,發現 Score 高分與 E value 為 0 的相同序列。
- 承上,這些 Score 高分與 E value 為 0 的相同序列表示為高度保留的序列區域。
- BLAST 操作 3 與結果:tblastx
將 yeast chromosome I 利用 tblastx 方式和 databases 中核酸序列進行比對時,會先將序列(先轉錄再)轉譯成蛋白質,然後根據蛋白質來進行比對,但是這時的 alignment 並沒有計算 gap,執行指令如下:
$ tblastx –db ./refseqgene1.gnome.fna –query ./chr01.fsa –out tblastx_out –evalue 0.000001 –num_threads 2
執行參考圖:

執行結果說明:
由結果可以看出,yeast chromosome I 與 refseqgene1.genomic.fna 經過 tblastx 比對後,發現兩者在 protein level 已有部分的相似度,如下;
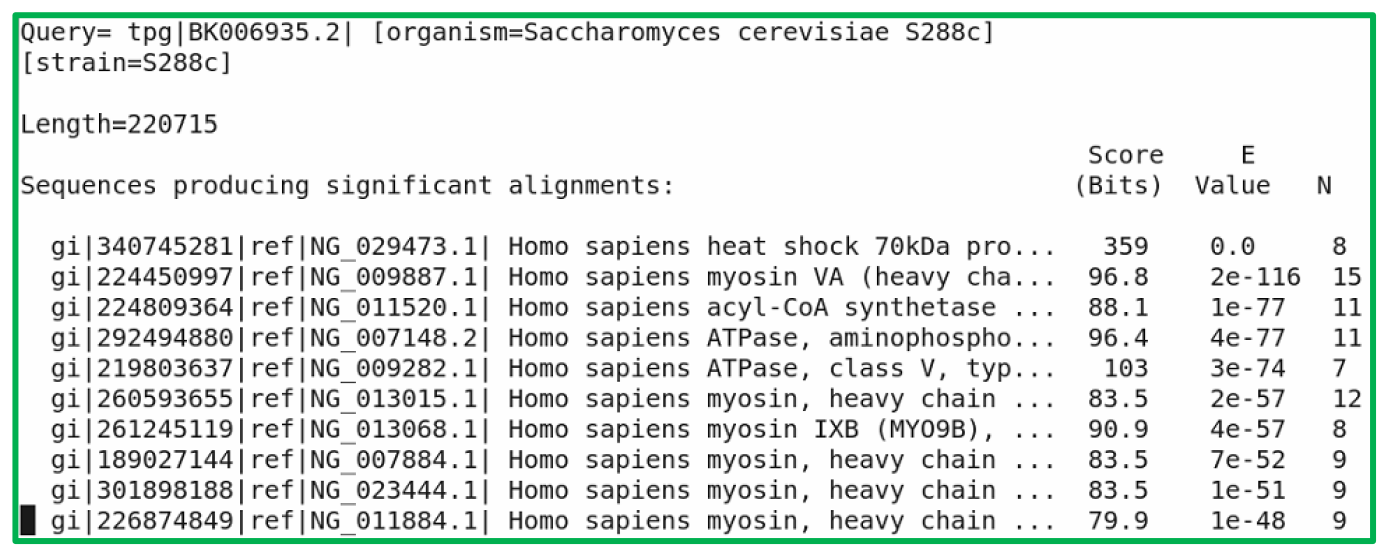
- Score(Bits):表示兩個序列的同源性,分數越高表示兩序列間相似程度越大。
- E value:表示在資料庫中找到與目標序列高相似度的可能性。和 S 值可以相互映襯,E 值應該越低越好,代表越不容易找到相同的序列。而 E value 算法如下:
$$E-value = K m n (e^{-\lambda S})$$
- $$ \lambda $$ 與數據庫和演算法有關,是一常數。
- m 表示目標序列的長度,n 表示資料庫的大小,S 即是前述的 Score 值。
- 常用 $$10^{-5}$$ 作為可比性結果。若 E = $$10^{-3}$$ 時,若有 1 個序列的 Score 前有 1000 條序列的分數更高,則當 E = $$10^{-6}$$ 時,則可能就僅會出現 1 個結果。
- E 值有侷限結果,如當目標序列較短,E 值會偏大,而且無法取得較高的 Scores,又如當兩序列同源性高,但有較大的 gap 時,S 值會下降,就必須依賴另一指標來取得 gap 的資訊,又如有些序列的 non-coding region 若是變異性較低,可能會造成這些序列的同源性較高, E value 值較高等。
- 因此對於 E value 來分析,當 E value 小於 $$10^{-5}$$ 時,表示兩序列有較高的同源性;若是 E value 小於 $$10^{-6}$$ ,表示兩序列的同源性很高。