Bowtie2 operation with RNA-seq data on windows
Download and Install
Download bowtie2 tool from its developing website [download], and take version 2.1.0 with 64-bit windows-based OS as a example.
Unzip (extract) the compressed files (name: bowtie2-2.1.0-mingw-win64.zip)
Move the folder already decompressed to location where your want to place, the following is an example ( C:\Useful Programs\ )
We further click the folder continuously to find the execution program (.exe), the following is a example:
C:\Useful Programs\bowtie2-2.1.0-mingw-win64\bowtie2-2.1.0Because tool bowtie2 is built and constructed on command line mode, we must use command line-like environment to run it. We next click cmd.exe or "命令提示字元".
And type the following format: (cd "the execution program folder location")
C:\> cd C:\Useful Programs\bowtie2-2.1.0-mingw-win64\bowtie2-2.1.0 # If the folder not in C disk, you would have to another type disk path as "disk:". For example: # C:\>D:
Basic operations
Following we would take basic operations with default examples to operate bowtie2 in command line mode. First of all, it is necessary to know parameters for bowtie2.
- the DNA-form sequence generated by next-generation sequencing (NGS) for further analyzing (testing): located on folder C:\Useful Programs\bowtie2-2.1.0-mingw-win64\bowtie2-2.1.0\example\reads . The file would be named as (sequence_name).fq format, e.g. reads_1.fq.
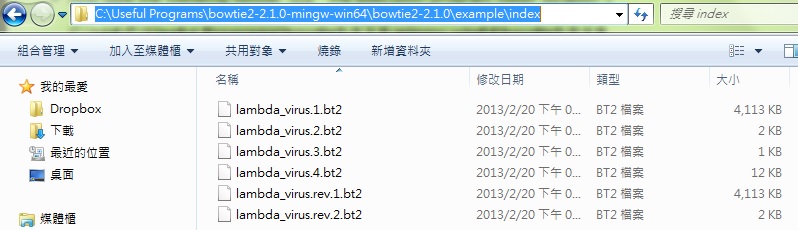
- the screening databases already indexed are used to align the testing sequences: located on folder C:\Useful Programs\bowtie2-2.1.0-mingw-win64\bowtie2-2.1.0\example\index . There might be several sub-files for a single alignment. For example, lambda_virus composes of six sub-files as the following image.
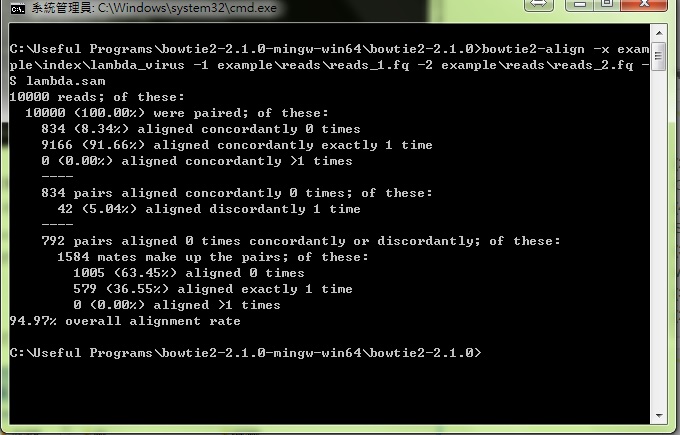
Next we try to align the NGS data with indexed databases in a simple following instruction.
C:\Useful Programs\bowtie2-2.1.0-mingw-win64\bowtie2-2.1.0>bowtie2-align -x example\index\lambda_virus -1 example\reads\reads_1.fq -2 example\reads\reads_2.fq -S lambda.sam- bowtie2-align : a execution program used to align the NGS data with indexed databases.
- -x : the basename of the index for the reference genome; the basename is the name specific to the front of a array of index-marked file names. For example, the basename of the array composed of lambda_virus.1.bt2, lambda_virus.2.bt2, lambda_virus.3.bt2, lambda_virus.4.bt2, etc. is lambda_virus.
- -1, -2 : The input sequence from NGS is paired and one strand is named with "_1" and the other is named with "_2" in general. When we use paired mate (/end) data. Parameters "-1" and "-2" represent each two separable data (strands). The parameter "-1" or "-2" could not be separately operated on bowtie2.
- -U : The input sequence from NGS is unpaired sequencing data.
- -S : Write a file in SAM format.
And the result is the following figure: