Phylogeny tree construction
- Case study: The extinction of the Tasmanian tiger
The thylacine
The Tasmanian tiger (scientific name: thylacine) was not a tiger but a dog-like marsupial animal.
It was a marsupial animal which the female thylacine may carry its young one in its pouch.
The thylacine resembled a much larger dog, such as the skull.
The thylacine was a predator and the largest known carnivorous marsupial in the modern age.
Extinction
The extinction of the thylacine resulted from two reasons:
- over-hunting of the human
- the inappropriate protection
For example, there were 3,482 Tasmanian tigers dispatched to London in order to produce waistcoats. ( cited from Owen D. (2003) Thylacine: The Tragic Tale of the Tasmanian Tiger. Sydney, Allen & Unwin. )
Recent DNA analysis
Although the appearance is similar between the thylacine and the dog, there are different evolution pathways of them. Such similarity between them is an example of convergent evolution, independent evolution pathways leading to the similar properties.
The skin of the thylacine was preserved and then as a DNA source was token to proceed sequencing of mitochondrial DNA. ( cited from Miller et al. (2009) The mitochondrial genome sequence of the Tasmanian tiger. Genome Res 19(2),213-220 )
The thylacine may be a good model for analysis of convergent evolution. How was the thylacine related to others marsupial species still present today ?
DNA amplification from two different thylacine animals:
- one sample: which comprised of two small pieces of attached hair in the early 20th century
- the other sample: a piece of dried muscle adhering to a bone and this sample was collected before 1983
the analysis of the complete mitochondrial genome sequence from two additional thylacine individuals:
- one sample: this one was the offspring of a female sent to the National Zoo in Washington, DC in 1902. (which died in 1905)
- the other sample: this one is the almost-complete animal stored in ethanol
Phylogeny tree construction - PHYLIP vs. ClustalW
The construction of the phylogenetic tree is based on several sequences of the mitochondrial genome, stored as a file "mito.fa".
This file containing many sequences of the mitochondrial genome:
- retrieved from NCBI Entrez system
- aligned with ClustalW
- removing columns with numerous gaps
- stored in the FASTA format
the several methods for the phylogeny tree construction:
- the neighbor-joining (refer to LINK) method of ClustalW: 1000 replicates used in the bootstrap
- the max parsimony analysis with the program Dnapars in the PHYLIP suit: 100 replicates used in the bootstrap
- MrBayes
the implement of neighbor-joining method of ClustalW on the Linux-based environment:
# the guide tree(similar with phylogeny tree) is stored as a file sequence.dnd in the same folder
$ ./clustalw2 ./sequence.fasta -bootstrap
- the implement of progeam Dnapars:
$ ./clustalw2 ./mito.fa -output=phylip
$ cp ./mito.phy infile
$ ./seqboot ./
$ cp ./outfile ./infile
$ ./dnapars ./
$ cp ./outtree ./intree
$ ./consense ./
PHYLIP :
- the PHYLIP suits and their instructions could be download: LINK
- use clustalw2 to generate the file for the suit phylip usage
- seqboot, dnapars and consense are the execution file stored in the folder exe
- both the file names infile and outfile are fixed so as to be further used in the next instructions
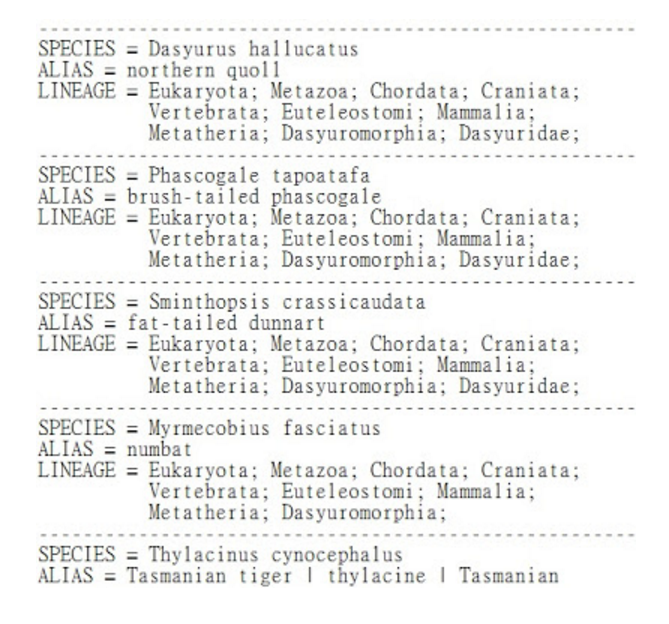
Capture the species lineage from Taxonomy database in Perl
input: a file, mito.fa, stores sequences in FASTA and a file, tax.txt, which the taxonomy of a hundred thousand of species
output: the complement information of each sequence in taxonomy file
the following is the implement
#!/usr/bin/perl -w
use strict;
# establish the database
my $libName = "tax.txt";
my %alias; # another name
my %lineage; # complete description
my @temp; # temp usage
my $count = 1;
open(fin,$libName) or die("The file tax.txt was wrong.");
foreach my $line () {
chomp($line);
@temp = split("\t",$line);
$alias{$temp[1]} = $temp[2]; # key is scientific name, value is GenBank common name
$lineage{$temp[1]} = $temp[3]; # key is scientific name, value is lineage information
}
close(fin);
# query the database
my $queryData = "mito.fa";
my $getSpecies = "";
my $getAlias = "";
my $getLineage = "";
# format designed
# self format design ends with '.'
# start with '@'
# each < represents a character which is left-justified
# '^' represents the filled field operator
# '~' prevents blank characters added to the end of the line and further to print
format =
------------------------------------------------------
SPECIES = @<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<< ~
$getSpecies
ALIAS = ^<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<< ~
$getAlias
^<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<< ~
$getAlias
LINEAGE = ^<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<< ~
$getLineage
^<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<< ~
$getLineage
^<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<< ~
$getLineage
.
open(fin,$queryData) or die("Query data was wrong.\n");
foreach my $line () {
chomp($line);
if($line =~ m/>/) {
$getSpecies = $line;
$getSpecies =~ s/_/ /g; # replace '_' with ' '
$getSpecies =~ s/>//g; # remove '>'
$getAlias = $alias{$getSpecies};
$getLineage = $lineage{$getSpecies};
write; # for format usage
}
}
close(fin);
- The partial result after the execution of the above script